








-
1
-
2
-
3
-
4
-
5
-
6
-
7
-
8
-
9
-
10
-
11
-
12
-
13
-
14
-
15
-
16
-
17
해당 자료는 5페이지 까지만 미리보기를 제공합니다.
5페이지 이후부터 다운로드 후 확인할 수 있습니다.
5페이지 이후부터 다운로드 후 확인할 수 있습니다.




목차
1. PRIDE (PRoteomics IDEntifications database)
2. wwPDB (World Wide Protein Data Bank)
3. Glycobiolgy & glycomics DB (from dkfz.)
4. PDB Server
5. Wikipedia : Protein Data Bank
2. wwPDB (World Wide Protein Data Bank)
3. Glycobiolgy & glycomics DB (from dkfz.)
4. PDB Server
5. Wikipedia : Protein Data Bank
본문내용
[52141]
4.Family: Uracil-DNA glycosylase [52142]
5.Protein: Uracil-DNA glycosylase [52143]
6.Species: Human (Homo sapiens) [52144]
7.domain : 1emh complexed with p2u; mutant
1.chain a [31008]
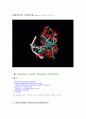
PDB ID : 1emh
Chain name : CRYSTAL STRUCTURE OF HUMAN URACIL-DNA GLYCOSYLASE BOUND TO UNCLEAVED SUBSTRATE-CONTAINING DNA
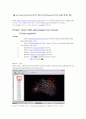
실제로 한 분자를 가리키니 protein structure상에서 해당되는 부분이 나온다.
atom 형식으로 관찰한 예
Line : atom(원모양)들과 atom사이 연결된 부분들이 선으로 표시되어지고 있다.
CPK : 모든 구성요소가 다 공모양으로 합쳐져 있는 듯한 모양을 보인다. 매우 복잡하다.
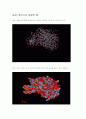
protein 형식으로 관찰한 예
soild-ribbon : atom을 없애고 protein의 연결 구조를 보기 위해 protein만 나타냈다.
α,β 구조(helix와 beta-strand)가 잘 나와 있다.
DNA 형식으로 관찰한 예
Backbone : arrows / Base Pairs : rings
전체적으로 나타낸 예 (protein이 복잡하게 나와 있다.)
Unknown-Protein Structure Prediction
Method
Secondary Structure Prediction
Solvent accessibility prediction & Folds
Transmembrane helix and signal peptide prediction
Prediction of coiled-coils
Prediction of O-glycosylation sites
Homology modelling
Threading
이런 방법들이 있다. 기본적인 원리는 4가지로 압축되어서 말할 수 있다.
1. Secondary structure prediction
multiple sequence을 alignment(정렬시켜서 분석)한 후, 그 결과를 본 다음에, 비슷한 indibidual sequence을 토대로 GOR method에 의해서 secondary structure을 알아내는 것이다. (75~80% 정확도)
2. Protein tertiary structure prediction
Target sequence을 가져다가 유사한 sequence를 찾는다. 이 유사한 sequence를 토대로 structure화 시켜서 본 것이 Temlate structure이다. 그래서 이 것과 target sequence를 비교해본다. 그럼으로써 target의 structure을 예상해 볼 수 있다. 이런 과정을 통해 structure을 입힌다. 그러나 나온 structure는 완벽하지는 않지만 비슷하다.
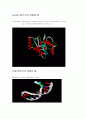
3. Threading & Fold recognition
Threading & Fold recognition은 target sequence에 structure를 각각 껴서 맞춰가는 것이다. 그래서 하나씩 해서 전체적으로 3차 구조를 만드는 것이다. 만약에 여러 가지 조합이 나왔다면, 확률을 따져서 최적의 조합으로 한다.
다음 그림은 이 Threading & Fold recognition이 어떻게 각각 sequence당 structure을 대입하게 되는지에 대한 설명을 나타내고 있다.
이러한 Protein Structure Prediction의 site로써 다음과 같이 나와 있다.
http://www.predictprotein.org/
Secondary Structure Prediction site
Name
Accuracy
Comments
PHDsec: http://www.embl-heidelberg.de/predictprotein/
> 72% (+/-10%, one standard deviation)
Multiple alignment-based neural network system.
NSSP: http://dot.imgen.bcm.tmc.edu:9331/pssprediction/pssp.html
> 71%. Evaluated on > 200 unique proteins.
Multiple alignment-based nearest-neigbour method.
SOPM: http://www.ibcp.fr/predict.html
> 70%.
Multiple alignment-based method combining various other prediction programs.
DSC: http://bonsai.lif.icnet.uk/bmm/dsc/dsc_read_align.html
70%
Multiple alignment-based program using statistics.
SSPRED: http://www.embl-heidelberg.de/sspred/ssp_mul.html
> 70%.
Multiple alignment-based program using statistics.
MultiPredict: http://kestrel.ludwig.ucl.ac.uk/zpred.html
> 65%
Multiple alignment-based method using physicochemical information from a set of aligned sequences and statistical secondary structure decision constants.
PSA: http://bmerc-www.bu.edu/psa/
The PSA server analyzes amino acid sequences to predict secondary structures and folding classes.
NNPREDICT: http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html
> 65%
Single-sequence based neural network prediction.
4.Family: Uracil-DNA glycosylase [52142]
5.Protein: Uracil-DNA glycosylase [52143]
6.Species: Human (Homo sapiens) [52144]
7.domain : 1emh complexed with p2u; mutant
1.chain a [31008]
PDB ID : 1emh
Chain name : CRYSTAL STRUCTURE OF HUMAN URACIL-DNA GLYCOSYLASE BOUND TO UNCLEAVED SUBSTRATE-CONTAINING DNA
실제로 한 분자를 가리키니 protein structure상에서 해당되는 부분이 나온다.
atom 형식으로 관찰한 예
Line : atom(원모양)들과 atom사이 연결된 부분들이 선으로 표시되어지고 있다.
CPK : 모든 구성요소가 다 공모양으로 합쳐져 있는 듯한 모양을 보인다. 매우 복잡하다.
protein 형식으로 관찰한 예
soild-ribbon : atom을 없애고 protein의 연결 구조를 보기 위해 protein만 나타냈다.
α,β 구조(helix와 beta-strand)가 잘 나와 있다.
DNA 형식으로 관찰한 예
Backbone : arrows / Base Pairs : rings
전체적으로 나타낸 예 (protein이 복잡하게 나와 있다.)
Unknown-Protein Structure Prediction
Method
Secondary Structure Prediction
Solvent accessibility prediction & Folds
Transmembrane helix and signal peptide prediction
Prediction of coiled-coils
Prediction of O-glycosylation sites
Homology modelling
Threading
이런 방법들이 있다. 기본적인 원리는 4가지로 압축되어서 말할 수 있다.
1. Secondary structure prediction
multiple sequence을 alignment(정렬시켜서 분석)한 후, 그 결과를 본 다음에, 비슷한 indibidual sequence을 토대로 GOR method에 의해서 secondary structure을 알아내는 것이다. (75~80% 정확도)
2. Protein tertiary structure prediction
Target sequence을 가져다가 유사한 sequence를 찾는다. 이 유사한 sequence를 토대로 structure화 시켜서 본 것이 Temlate structure이다. 그래서 이 것과 target sequence를 비교해본다. 그럼으로써 target의 structure을 예상해 볼 수 있다. 이런 과정을 통해 structure을 입힌다. 그러나 나온 structure는 완벽하지는 않지만 비슷하다.
3. Threading & Fold recognition
Threading & Fold recognition은 target sequence에 structure를 각각 껴서 맞춰가는 것이다. 그래서 하나씩 해서 전체적으로 3차 구조를 만드는 것이다. 만약에 여러 가지 조합이 나왔다면, 확률을 따져서 최적의 조합으로 한다.
다음 그림은 이 Threading & Fold recognition이 어떻게 각각 sequence당 structure을 대입하게 되는지에 대한 설명을 나타내고 있다.
이러한 Protein Structure Prediction의 site로써 다음과 같이 나와 있다.
http://www.predictprotein.org/
Secondary Structure Prediction site
Name
Accuracy
Comments
PHDsec: http://www.embl-heidelberg.de/predictprotein/
> 72% (+/-10%, one standard deviation)
Multiple alignment-based neural network system.
NSSP: http://dot.imgen.bcm.tmc.edu:9331/pssprediction/pssp.html
> 71%. Evaluated on > 200 unique proteins.
Multiple alignment-based nearest-neigbour method.
SOPM: http://www.ibcp.fr/predict.html
> 70%.
Multiple alignment-based method combining various other prediction programs.
DSC: http://bonsai.lif.icnet.uk/bmm/dsc/dsc_read_align.html
70%
Multiple alignment-based program using statistics.
SSPRED: http://www.embl-heidelberg.de/sspred/ssp_mul.html
> 70%.
Multiple alignment-based program using statistics.
MultiPredict: http://kestrel.ludwig.ucl.ac.uk/zpred.html
> 65%
Multiple alignment-based method using physicochemical information from a set of aligned sequences and statistical secondary structure decision constants.
PSA: http://bmerc-www.bu.edu/psa/
The PSA server analyzes amino acid sequences to predict secondary structures and folding classes.
NNPREDICT: http://www.cmpharm.ucsf.edu/~nomi/nnpredict.html
> 65%
Single-sequence based neural network prediction.
추천자료
- 가격1,900원
- 페이지수17페이지
- 등록일2007.07.29
- 저작시기2007.6
- 파일형식한글(hwp)
- 자료번호#423228

본 자료는 최근 2주간 다운받은 회원이 없습니다.